液相色谱实验报告

华南师范大学实验报告
液相色谱分析混合样品中的苯和甲苯
一、实验目的
1、掌握高效液相色谱定性和定量分析的原理及方法;
2、
了解高效液相色谱的构造、原理及操作技术。
二、实验原理
高效液相色谱由储液器,泵、进样器、色谱柱、检测器、记录仪等几部分组成,储液器中的流动相被高压泵打入系统,样品溶液经进样器进入流动相,被流动相载入色谱柱内,由于样品溶液中的各组分在两相中具有不同的分配系数,在两相中作相对运动,经过反复多次的吸附—解吸的分配过程,各组分在移动速度上产生较大的差别,被分离成单个组分依次从柱内流出,通过检测器时,样品浓度被转换成电信号传送到记录仪。
三、主要仪器和试剂
主要仪器:岛津液相色谱仪(LC-10AT)[配有紫外检测器,Phenomenex ODS 柱];10μL 微量注射器
试剂:苯标准溶液:10.0μL/mL;
甲苯标准溶液:10.0μL/mL;
苯、甲苯混合标准溶液:10.0μL/mL;
甲醇:80% ;
苯和甲苯混合待测溶液;
四、实验步骤
1、标准溶液的配制系列
用100μL的微量注射器分别量取10μL、20μL、50μL、100μL的苯和甲苯的混合标准溶液(10.0μL/mL),再分别加入90μL、80μL、50μL、0μL甲醇将其稀释,作为待测液,其浓度分别为1μL/mL、2μL/mL、5μL/mL、10μL/mL
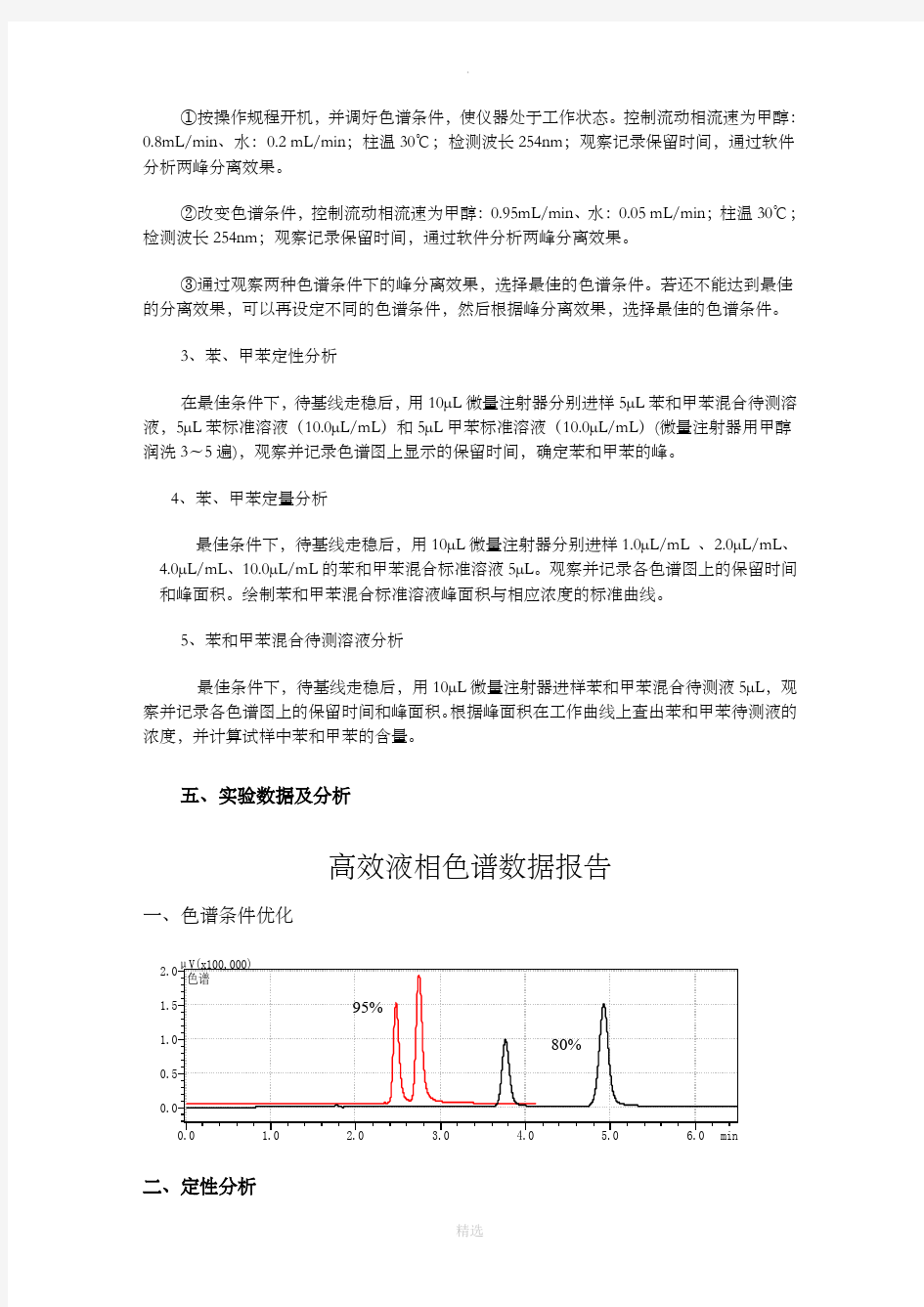
2、色谱条件优化
①按操作规程开机,并调好色谱条件,使仪器处于工作状态。控制流动相流速为甲醇:0.8mL/min 、水:0.2 mL/min ;柱温30℃;检测波长254nm ;观察记录保留时间,通过软件分析两峰分离效果。
②改变色谱条件,控制流动相流速为甲醇:0.95mL/min 、水:0.05 mL/min ;柱温30℃;检测波长254nm ;观察记录保留时间,通过软件分析两峰分离效果。
③通过观察两种色谱条件下的峰分离效果,选择最佳的色谱条件。若还不能达到最佳的分离效果,可以再设定不同的色谱条件,然后根据峰分离效果,选择最佳的色谱条件。
3、苯、甲苯定性分析
在最佳条件下,待基线走稳后,用10μL 微量注射器分别进样5μL 苯和甲苯混合待测溶液,5μL 苯标准溶液(10.0μL/mL)和5μL 甲苯标准溶液(10.0μL/mL)(微量注射器用甲醇润洗3~5遍),观察并记录色谱图上显示的保留时间,确定苯和甲苯的峰。
4、苯、甲苯定量分析
最佳条件下,待基线走稳后,用10μL 微量注射器分别进样1.0μL/mL 、2.0μL/mL、4.0μL/m L 、10.0μL/mL 的苯和甲苯混合标准溶液5μL。观察并记录各色谱图上的保留时间和峰面积。绘制苯和甲苯混合标准溶液峰面积与相应浓度的标准曲线。 5、苯和甲苯混合待测溶液分析
最佳条件下,待基线走稳后,用10μL 微量注射器进样苯和甲苯混合待测液5μL,观察并记录各色谱图上的保留时间和峰面积。根据峰面积在工作曲线上查出苯和甲苯待测液的浓度,并计算试样中苯和甲苯的含量。
五、实验数据及分析
高效液相色谱数据报告
一、色谱条件优化
0.0
1.0
2.0
3.0
4.0
5.0
6.0
min
0.00.51.01.52.0μV (x100,000)
色谱
二、定性分析
95%
80%
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5min
0.01.02.03.04.05.0μV
(x10,000)
色谱
三、 定量分析
苯、甲苯混合标准溶液浓度分别为:1 ul/ml, 2 ul/ml, 5 ul/ml, 10 ul/ml 。
苯
甲苯
1 ul/ml
10 ul/ml
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
min
0.00.51.01.52.02.53.03.54.0μV (x100,000)
色谱
四、未知样品浓度
六、实验讨论
①由于实验中待测溶液的组分只有苯和甲苯两种物质,组分比较简单,只要在实验过程中,严格控制且定量进样,就能采用操作、计算简便的标准曲线法得出准确的实验结果,
因而采用标准曲线法。若采用归一化法或内标法,虽然能得到准确更高的实验结果,但由于实验操作及计算均较为复杂,实验及数据处理的时间长,效率相对不高,故实验中采用标准曲线法作为定量的方法。
②最佳色谱条件的选择是本实验准确度的关键。若苯与甲苯的峰分离效果过小,苯与甲苯的峰未能完全分离,必定影响其保留时间和峰面积的计算,是实验准确度减小,数据误差加大;若苯与甲苯的峰分离效果过好,虽然能将苯与甲苯的峰完全分离,实验准确度好,但是耗费的实验时间较长,效率不好。因而,应选择合适的色谱条件。
③为了提高标准曲线的拟合度,可以重复进样多次,并且由同一人进样,以免因个体习惯不同引起实验误差。进样针进样时确保进样针中没有残留气泡,另外,在进不同浓度或不同的物质前因先用乙醇溶液润洗干净,以免相互影响。
高效液相色谱法简介
高效液相色谱法简介 “色谱”一词是由俄国科学家斯威特提出的。色谱法是基于补充物质在相对运动物的两相之间分布时,物理或物理化学性质的微小的差异而使混合物相互分离的一类分离或分析方法。发展与上世纪初,飞速发展于五十年代,有超过30位科学家家因为它而获得诺贝尔奖,其有自己的理论和研究方法,同时也有众多的应用领域。 色谱法常见的方法有:柱色谱法、薄层色谱法、气相色谱法、高效液相色谱法等。 柱色谱:柱色谱法是最原始的色谱方法,这种方法将固定相注入下端塞有棉花或滤纸的玻璃管中,将被样品饱和的固定相粉末摊铺在玻璃管顶端,以流动相洗脱。常见的洗脱方式有两种,一种是自上而下依靠溶剂本身的重力洗脱,一种是自下而上依靠毛细作用洗脱。收集分离后的纯净组分也有两种不同的方法,一种方法是在柱尾直接接受流出的溶液,另一种方法是烘干固定相后用机械方法分开各个色带,以合适的溶剂浸泡固定相提取组分分子。柱色谱法被广泛应用于混合物的分离,包括对有机合成产物、天然提取物以及生物大分子的分离。 薄层色谱:薄层色谱法是应用非常广泛的色谱方法,这种色谱方法将固定相图布在金属或玻璃薄板上形成薄层,用毛细管、钢笔或者其他工具将样品点染于薄板一端,之后将点样端浸入流动相中,依靠毛细作用令流动相溶剂沿薄板上行展开样品。薄层色谱法成本低廉操作简单,被用于对样品的粗测、对有机合成反应进程的检测等用途。
气相色谱:GC主要是利用物质的沸点、极性及吸附性质的差异来实现混合物的分离。待分析样品在汽化室汽化后被惰性气体(即载气,也叫流动相)带入色谱柱,柱内含有液体或固体流动相,由于样品中各组分的沸点、极性或吸附性能不同,每种组分都倾向于在流动相和固定相之间形成分配或吸附平衡。但由于载气是流动的,这种平衡实际上很难建立起来。也正是由于载气的流动,使样品组分在运动中进行反复多次的分配或吸附/解吸附,结果是在载气中浓度大的组分先流出色谱柱,而在固定相中分配浓度大的组分后流出。当组分流出色谱柱后,立即进入检测器。检测器能够将样品组分的与否转变为电信号,而电信号的大小与被测组分的量或浓度成正比。当将这些信号放大并记录下来时,就是气相色谱图了。气相色谱被广泛应用于小分子量复杂组分物质的定量分析。 高效液相色谱:高效液相色谱法是在经典色谱法的基础上,引用了气相色谱的理论,在技术上,流动相改为高压输送(最高输送压力可达4.9-107Pa);色谱柱是以特殊的方法用小粒径的填料填充而成,从而使柱效大大高于经典液相色谱(每米塔板数可达几万或几十万);同时柱后连有高灵敏度的检测器,可对流出物进行连续检测。高效液相色谱(HPLC)是目前应用最多的色谱分析方法,高效液相色谱系统由流动相储液体瓶、输液泵、进样器、色谱柱、检测器和记录器组成,其整体组成类似于气相色谱,但是针对其流动相为液体的特点作出很多调整。HPLC的输液泵要求输液量恒定平稳;进样系统要求进样便利切换严密;由于液体流动相粘度远远高于气体,为了减低柱压高效
气相色谱法实验报告记录
气相色谱法实验报告记录
————————————————————————————————作者:————————————————————————————————日期:
实验五—气相色谱法实验 姓名:张瑞芳 学号:2013E8003561147 班级:化院413班 培养单位:上海高等研究院 指导教师:李向军 组别:2013年12月30日第二组
气相色谱法实验 一、实验目的 1.了解气相色谱仪的各部件的功能。 2.加深理解气相色谱的原理和应用。 3.掌握气相色谱分析的一般实验方法。 4.学会使用FID气相色谱对未知物进行分析。 二、实验原理 1.气相色谱法基本原理 气相色谱的流动向为惰性气体,气-固色谱法中以表面积大且具有一定活性的吸附剂作为固定相。当多组分的混合样品进入色谱柱后,由于吸附剂对每个组分的吸附力不同,经过一定时间后,各组分在色谱柱中的运行速度也就不同。吸附力弱的组分容易被解吸下来,最先离开色谱柱进入检测器,而吸附力最强的组分最不容易被解吸下来,因此最后离开色谱柱。如此,各组分得以在色谱柱中彼此分离,顺序进入检测器中被检测、记录下来。气相色谱仪器框图如图1所示: 图1.气相色谱仪器框图 仪器均由以下五个系统组成:气路、进样、分离、温度控制、检测和记录系统。 2.气相色谱法定性和定量分析原理 在这种吸附色谱中常用流出曲线来描述样品中各组分的浓度。也就是说,让
分离后的各组分谱带的浓度变化输入换能装置中,转变成电信号的变化。然后将电信号的变化输入记录器记录下来,便得到如图2的曲线。它表示组分进入检测器后,检测器所给出的信号随时间变化的规律。它是柱内组分分离结果的反映,是研究色谱分离过程机理的依据,也是定性和定量的依据。 图2.典型的色谱流动曲线 3.FID的原理 本次试验所用的为氢火焰离子化检测器(FID),它是以氢气和空气燃烧的火焰作为能源,利用含碳有机物在火焰中燃烧产生离子,在外加的电场作用下,使离子形成离子流,根据离子流产生的电信号强度,检测被色谱柱分离出的组分。 三.实验试剂和仪器 (1)试剂:甲醇、异丙醇、异丁醇 (2)仪器:气相色谱仪带氢火焰离子化检测器(GC-2014气相色谱仪); 氢-空发生器(SPH-300氢气发生器)、氮气钢瓶; 色谱柱; 微量注射器。 四.实验步骤 1.打开稳定电源。 2.打开N2钢瓶(减压阀),以N2为载气,开始通气,检漏;调整柱前压约为 0.12MPa。
液相色谱分析混合样品中的苯和甲苯
华南师范大学实验报告 课程名称仪器分析实验实验项目液相色谱分析混合样品中的苯和甲苯 实验类型□验证□设计□综合实验时间2010 年 3 月31 日 实验指导老师实验评分 一、实验目的 1.掌握高效液相色谱定性和定量分析的原理及方法; 2.了解高效液相色谱的构造、原理及操作技术。 二、实验原理 高效液相色谱由储液器,泵、进样器、色谱柱、检测器、记录仪等几部分组成。储液器中的流动相被高压泵打入系统,样品溶液经进样器进入流动相,被流动相载入色谱柱内。由于样品溶液中的各组分在两相中具有不同的分配系数,在两相中作相对运动是,经过反复多次的吸附-解吸的分配过程,各组分在移动速度上产生较大的差别,被分离成单个组分依次从柱内流出,通过检测器时,样品浓度被转换成电信号传送到记录仪,记录成数据。 液相色谱的定性依据是保留时间的相对性,通常相对误差不能大于5%。定量参数常常采用峰高、峰面积、相对峰高、相对峰面积等。定量方法通常采用外标法、内标法和面积归一化法。外标法为标准物质标准溶液制定标准曲线法;内标法为在标准溶液、样品溶液中加入内标物质,以相对峰高、相对面积对标准物质的浓度制定标准曲线;面积归一化法假定所有出峰物质的吸光系数相同,计算某物质的峰面积占所有峰面积的百分比。 三、仪器与试剂: 1.仪器:SCL-10A vp紫外可见双波长检测器;SPD-M10A vp柱温箱;LC-10AT高效液相
色谱仪;10μL微量注射器 2.试剂:2μL/mL苯标液;2μL/mL甲苯标液;0.2μL/mL、2μL/mL、4μL/mL、10μL/mL 的苯与甲苯的混合标准溶液;甲醇溶液;待测试样溶液 四、实验内容与步骤: 1.选择合适的流动相配比,优化色谱条件 设置有关参数:控制流速为1mL/min。柱温30℃,检测波长354nm。 设置流动相配比(甲醇:水=1:1),用10μL微量注射器注射5μL的10μL/mL苯与甲苯的混合标准溶液进行测定,观察其分离度和出峰时间。然后改变流动相配比,改进分离,调整出峰时间。从而得到最佳测定条件。 2.苯、甲苯定性分析: 在最佳的测定条件下,用10μL微量注射器,分别注射5μL2μL/mL苯的标准溶液和2μL/mL甲苯的标准溶液。观察记录保留时间,确定苯和甲苯的峰。 3.苯、甲苯定量分析: 在最佳的测定条件下,用10μL微量注射器,分别注射5μL 0.2μL/mL、2μL/mL、4μL/mL、10μL/mL苯与甲苯的混合标准溶液,再分别测定苯和甲苯的峰面积,以峰面积对浓度作图,做出工作曲线。 在最佳的测定条件下,用10μL微量注射器,注射5μL的试样,观察记录保留时间和峰面积。根据峰面积在工作曲线上查处苯和甲苯待侧溶液的浓度,并计算试样中苯和甲苯的含量。 五、数据记录及结果分析: 1. 优化色谱条件
高效液相色谱实验报告
高效液相色谱实验报告 一、实验目的 1了解液相色谱的发展历史及最新进展 2 学习液相色谱的基本构造及原理 3 掌握液相色谱的操作方法和分析方法,能够通过HPLC分离测定来对目标化合物的分析鉴定。 二、实验原理 液相色谱法采用液体作为流动相,利用物质在两相中的吸附或分配系数的微小差异达到分离的目的。当两相做相对移动时,被测物质在两相之间进行反复多次的质量交换,使溶质间微小的性质差异产生放大的效果,达到分离分析和测定的目的。液相色谱与气相色谱相比,最大的优点是可以分离不可挥发而具有一定溶解性的物质或受热后不稳定的物质,这类物质在已知化合物中占有相当大的比例,这也确定了液相色谱在应用领域中的地位。 高效液相色谱可分析低分子量、低沸点的有机化合物,更多适用于分析中、高分子量、高沸点及热稳定性差的有机化合物。80%的有机化合物都可以用高效液相色谱分析,目前以已经广泛应用于生物工程、制药工程、食品工业、环境检测、石油化工等行业。 三、高效液相色谱的分类 吸附色谱法、分配色谱法、空间排阻色谱法、离子交换色谱法、亲和色谱法、化学键合相色谱法 四、高效液相色谱仪的基本构造 高效液相色谱至少包括输液系统、进样器、分离柱、检测器和数据处理系统等几部分。 1 输液系统: 包括贮液及脱气装置、高压输液泵和梯度洗脱装置。贮液装置用于存贮足够量、符合HPLC要求的流动相。高效液相色谱柱填料颗粒比较小,通过柱子的流动相受到的流动阻力很大,因此需要高压泵输送流动相。 2 进样系统: 将待测的样品引入到色谱柱的装置。液相色谱进样装置需要满足重复性好、死体积小、保证柱中心进样、进样时引起的流量波动小、便于实现自动化等多项要求。进样系统包括取样、进样两项功能。 3 分离柱: 色谱柱是色谱仪的心脏、柱效高、选择性好、分析速度快是对色谱柱的一般要求。商品化的HPLC微粒填料,如硅胶和以硅胶为基质的键合相、氧化铝、有机聚合物微球(包括离子交换树脂)等的粒度通常在3μm、5μm、7μm、以及10μm。采用的固定相粒度甚至可以达到1μm,而制备色谱所采用的固定相粒度通常大于10μm。HPLC填充柱效的理论值可以达到50000/m~160000/m理论板,一般采用100-300mm的柱长可满足大多数样品的分析的需要。由于柱效内、外多种因素的影响,因此为使色谱柱达到其应有的效率。应尽量的减小系统的死体积。 4 检测系统: HPLC检测器分为通用型检测器和专用型检测器两类。通用型检测器可连续测量色谱柱流出物(包括流动相和样品组分)的全部特性变化。这类检测仪器包括示差折光检测器、介
HPLC实验高效液相色谱分析实验
仪器分析实验报告实验名称:高效液相色谱分析实验
一、实验目的 1. 了解HPLC的结构,了解仪器的开、关程序。 2. 了解流动相的制备和样品溶液的制备。 3. 知道仪器的运行程序和进行样品分析。 二、仪器和试剂 仪器:美国安捷伦1200型HPLC、10μL的微量注射器 试剂:磷酸乙腈溶液(PH=3)、重蒸水、邻氯苯甲酸 三、实验步骤 1.流动相的准备 流动相只有一组:PH=3的磷酸乙腈溶液,进过脱气,用蠕动泵输送。2.开机,色谱柱平衡 当1完成后,开机,待色谱柱平衡。 3.样品溶液的准备 配置好邻氯苯甲酸溶液,按要求选好滤纸的孔径大小。用低压过滤装置过滤,由于美国安捷伦1200型HPLC配有脱气装置,因此滤液无需事先脱气就可以进行分析。 4.基线的查看 由于仪器内部压力的变化可以引起基线的不断波动,因此,需等待压力稳定后,基线平稳才能进行进样。 5.样品进样分析
用10μL的微量注射器取5μL的邻氯苯甲酸,微量注射器中不能有气泡,将微量注射器的针头插入到注射的孔时,打开微量注射阀,将邻氯苯甲酸注射进去后,迅速关闭阀门,抽出针头,等待仪器的分析结果。 6.色谱柱的清洗 分析工作结束后,要清洗进样阀中的残留样品,也要用适当的液体来清洗色谱柱。 7.关机 实验完毕后,关闭仪器和电脑。 四、实验数据及处理 1.LC参数 2.色谱柱参数 3.四元泵状态 A:0.0%流速:1.000ml/min B:0.0%压力:91bar C:0.0% D:0.0%
5.色谱分析谱图见附页,经过注射5μL的邻氯苯甲酸,得到三组实验色谱图, 根据谱图列表数据如下: 色谱柱长(L)、理论塔板高度(H)与理论塔板数(n)三者的关系为: n = L / H 理论塔板数和色谱参数之间的关系为: n = 16 ( t R / W b ) 2 = 5.54 ( t R / Y1/2 ) 2 则取第五组数据计算得: t R=2.437 min = 146.22s Y1/2 = 2.354(0.1375min / 4 ) = 4.855125 s n = 5.54 ( t R / Y1/2 ) 2 =5025 (块)
液相色谱实验报告
华南师范大学实验报告 液相色谱分析混合样品中的苯和甲苯 一、实验目的 1、掌握高效液相色谱定性和定量分析的原理及方法; 2、了解高效液相色谱的构造、原理及操作技术。 二、实验原理 高效液相色谱由储液器,泵、进样器、色谱柱、检测器、记录仪等几部分组成,储液器中的流动相被高压泵打入系统,样品溶液经进样器进入流动相,被流动相载入色谱柱内,由于样品溶液中的各组分在两相中具有不同的分配系数,在两相中作相对运动,经过反复多次的吸附—解吸的分配过程,各组分在移动速度上产生较大的差别,被分离成单个组分依次从柱内流出,通过检测器时,样品浓度被转换成电信号传送到记录仪。 三、主要仪器和试剂 主要仪器:岛津液相色谱仪(LC-10AT)[配有紫外检测器,Phenomenex ODS 柱]; 10μL微量注射器 试剂:苯标准溶液:10.0μL/mL; 甲苯标准溶液:10.0μL/mL; 苯、甲苯混合标准溶液:10.0μL/mL; 甲醇:80% ; 苯和甲苯混合待测溶液; 四、实验步骤 1、标准溶液的配制系列 用100μL的微量注射器分别量取10μL、20μL、50μL、100μL的苯和甲苯的混合标准溶液(10.0μL/mL),再分别加入90μL、80μL、50μL、0μL甲醇将其稀释,作为待测液,其浓度分别为1μL/mL、2μL/mL、5μL/mL、10μL/mL 2、色谱条件优化 ①按操作规程开机,并调好色谱条件,使仪器处于工作状态。控制流动相流速为甲醇: 0.8mL/min、水:0.2 mL/min;柱温30℃;检测波长254nm;观察记录保留时间,通过软件分析两峰分离效果。 ②改变色谱条件,控制流动相流速为甲醇:0.95mL/min、水:0.05 mL/min;柱温30℃;检测波长254nm;观察记录保留时间,通过软件分析两峰分离效果。
高效液相色谱法备课资料
第二十章高效液相色谱法 Chapter 20 High performance liquid chromatography 本章讨论高效液相色谱法的主要类型和原理、固定相和流动相及其选择、高效液相色谱仪、高效液相色谱分析方法。本次课将重点讨论前半部分的内容,并注重与常规液相色谱、薄层色谱以及气相色谱的比较复习。 本章的结构编排比气相色谱法合理、流畅。 方法基本构架:经典LC【1964年Giddings发明了高效液相色谱法】 理论参考:GC 传统TLC: 1、缺乏强劲驱动力,达不到“HP”; 2、重现性较差; 3、缺乏通用灵敏的检测器; 4、由此造成的研究、生产投入的不足。 仪器化、自动化差,普及认知率相对低,恶性循环 HPLC: 1、以高压泵产生强劲驱动力,有了“HP”的基础; 2、ODS的问世(50%以上的应用) 3、仪器化、自动化,认知程度不断提高,良性循环(导致更多的优良的SP 的诞生、更灵敏更强大适用面更广的检测器的诞生、仪器性能更稳定更智能)HPLC cf经典LC 1、以高压泵产生强劲驱动力,快速高效(分钟计/小时计) 2、SP颗粒细、均匀,C项小,柱效高(<10um/>100um) *:TLC都是“离线检测”的方法,结果较粗,效率较低。 GC都是“在线检测”的方法,结果较精密,效率较高。 3、在线检测器种类更多、更灵敏、功能更强大、适用面更广 HPLC cf GC 1、适用样品种类多(80%的有机化合物) 【2、柱效、分析速度稍逊于GC】 3、不受试样挥发性低、热稳定性差的限制 4、HPLC选择MP的余地相对大,GC选择SP的余地相对大 目前HPLC已发展为分析领域一项最重要的分离分析方法,涉及各大行业的各类样品。在药学领域中,在生物样品、中药等复杂体系分析等方面举足轻重!
高效液相色谱法测定邻苯二甲酸酯实验报告记录
高效液相色谱法测定邻苯二甲酸酯实验报告记录
作者: 日期: 2
高效液相色谱法测定邻苯二甲酸酯 1553607胡艺蕾 实验时间:2017年4月1日实验温度:19.0 C 、实验目的 1、了解高效液相色谱仪的组成及其工作原理和基本操作。 2、对邻苯二甲酸酯进行分离和测定。 3、探究不同流动相及不同流动相比例对流速、柱压、保留时间及分离度的影响。 4、了解液相色谱法定量测定的原理。 二、实验原理 1、实验采用的反相液固吸附色谱法,其分离机理是:当流动相通过吸附剂时,在吸附剂(固体相)表面发生了溶质分子取代吸附剂上的溶剂分子的吸附作用。固体相为非极性分子,如十八烷基键合相,流动相为极性分子。 2、组分分子与吸附剂之间作用力的强弱决定它的保留时间。溶质分子官能团的性质和分子结构的空间效应都会影响其出峰的顺序。本次实验为邻苯二甲酸酯,其分子官能团都相同, 但由于DMP其官能团相邻的烷基较小,导致其保留值最小,因此出峰顺序为:DMP(邻苯二甲酸二甲酯)>DEP邻苯二甲酸二乙酯)>DBP(邻苯二甲酸二丁酯)。 3、在吸附色谱中,流动相的洗脱能力与溶剂的极性有关,极性越大,洗脱强度也越大。本次实验使用的三个流动相的极性大小为:水>乙腈>甲醇。通常选择二元混合溶剂作为流动相。 4、定量分析中,定量峰与其他峰之间的分离程度称为分离度 R: 通常用塔板数n来描述色谱的柱效: 三、实验仪器与试剂 1、仪器 Agilent1260高效液相色谱仪:
脱气机:真空室内半透膜管路,对流动相进行脱气四元泵:二元泵各控制一种溶剂可设置的流速范围:0.001 - 10 mL/min 0.001 mL/min 步进UV检测器:用于检测通过样品后的紫外光 类型:双光束光路设计 光源:氘灯波长范围:190 - 600 nm 手动进样器:进样20L 色谱柱:填料汁八烷(适合中性、弱酸碱) 4.6x 100mm, 3.5 1 m 2、试剂 流动相:纯水、甲醇、乙腈 样品:DMP、DEP DBP 四、实验步骤 1、开启电脑,开启脱气机、泵、检测器等的电源,启动软件。 2、预先脱气(直到导管中无气泡),设定波长:220nm。 3、设定流速、流动相比例等参数,选择合适的流动相。 4、进样阀柄置于“ LOAD',进样针用乙醇洗涤2-3次,取样,进样,将进样阀扳至 5、保存并处理数据。 五、实验结果 1、样品:DMP1:20水溶液201 L流动相的比例为:高纯水:30%乙腈:70%流速: “INJECT。 1.00ml/min 4
实例解析——高效液相色谱(hplc)
实例解析——高效液相色谱(HPLC) 一、原理 利用不同物质在两相中(液液、液固、离子交换、尺寸排阻)具有不同的分配系数,当二者相对运动时候,物质在两相中反复多次分配,从而使得物质得到完全分离 二、适用范围 高沸点、热不稳定的天然产物、生物大分子、高分子化合物、离子型样品、生化样品三、特点 高压、高效、高灵敏度 四、仪器组成 流动液贮存提供脱气,输液系统、进样系统、分离系统、检测系统,控制记录系统贮液瓶、高压泵、进样器、分离柱、检测器、记录仪 五、仪器选择 由实验条件确定是选用二元高压还是四元低压、一般来说,二元高压的准确度较高。四元低压是先将样品按比例混合再泵入,而二元高压是先泵入不同比例的溶剂再混合。确定采用的脱气系统,一般采用在线脱气。确定进样方式,人工手动六通阀进样,还是进样针自动进样,一个适用于少量样品,一个适用于大量样品。 选择检测器,如果是有较强的紫外吸收的可用紫外可见检测器(二极管阵列检测器),如果是芳香族化合物,可选用荧光检测器,对于离子可采用电导检测器。 六、实验条件优化 配置待测物质的标准溶液 1、色谱柱的确定 分析样本确定是采用何种类型的色谱柱 (1)分配色谱,两项间分配系数 流动相选用极性的物质(甲醇、乙腈、水)则固定相选择非极性物质。一般用 C18 ODS柱。 (2)吸附色谱, (3)离子交换色谱 各种离子与树脂上交换集团的交换能力不同。固定相:离子交换树脂,流动相 为无机酸、无机碱。常用于分离离子或者可解离的化合物 (4)排阻色谱法 配置含待测物质的标准品溶液,采用不同C18柱分离,检测,对照不同色谱图像,可得到分离效能最高的色谱柱 2、最佳流动相梯度洗脱程序的确定 梯度洗脱:按照一定的程度,不断改变流动相中个溶剂组成的比例以改变流动相的 极性。将色谱柱上不同的组分洗脱出来。 配置不同的梯度洗脱方案,用标准溶液进行试验,并选取能达到最高分离效能的梯 度洗过方案作为最佳流动相梯度洗脱程序 3、流动相的确定 在分离效能相似条件下选择更经济、毒性小的流动相 4、流速确定 流速太大,待分离组分来不及与固定相充分作用,故其中的组分较易被洗脱下来,出峰时间变短,而且柱压比较高,会引起泵负荷的增加,进而导致色谱柱的使用命
液相色谱实验报告
液相色谱实验报告 姓名:XXX 专业:有机化学学号:312070303004 时间:2012.11.11 一、实验目的: 1 .了解液相色谱仪的基本构造、工作原理。 2. 掌握液相色谱的操作方法和分析方法,能够通过高效液相色谱(HPLC)对目标化合物进行分析鉴定。 二、实验原理: 液相色谱法采用液体作为流动相,利用物质在两相中的吸附或分配系数的微小差异达到分离的目的。当两相做相对移动时,被测物质在两相之间进行反复多次的质量交换,使溶质间微小的性质差异产生放大的效果,达到分离分析和测定的目的。被分离成单个组分后依次从柱内流出,通过检测器时,样品浓度被转换成电信号传送到记录仪。 三、液相色谱法的特点: 高压——压力可达150~300 kg/cm2。色谱柱每米降压为75 kg/cm2以上。 高速——流速为0.1~10.0 mL/min。 高效——塔板数可达5000/米。在一根柱中同时分离成份可达100种。 高灵敏度——紫外检测器灵敏度可达0.01ng。同时消耗样品少。 高效液相色谱(HPLC)与经典液相色谱相比有以下优点: 速度快——通常分析一个样品在15~30 min,有些样品甚至在5 min内即可完成。 分辨率高——可选择固定相和流动相以达到最佳分离效果。 灵敏度高——紫外检测器可达0.01ng,荧光和电化学检测器可达0.1pg。 色谱柱可反复使用——用一根色谱柱可分离不同的化合物。 样品量少,容易回收——样品经过色谱柱后不被破坏,可以收集单一组分或做制备。 四、液相色谱仪的组成: 液相色谱仪主要包括输液系统、进样器、分离柱、检测器和数据处理系统等几部分。 1 输液系统: 包括贮液及脱气装置、高压输液泵和梯度洗脱装置。贮液装置用于存贮足够量、符合HPLC要求的流动相。高效液相色谱柱填料颗粒比较小,通过柱子的流动相受到的流动阻力很大,因此需要高压泵输送流动相。 2 进样系统: 将待测的样品引入到色谱柱的装置。液相色谱进样装置需要满足重复性好、死体积小、保证柱中心进样、进样时引起的流量波动小、便于实现自动化等多项要求。进样系统包括取样、进样两项功能。 3 分离柱: 色谱柱是色谱仪的心脏、柱效高、选择性好、分析速度快是对色谱柱的一般要求。商品化的HPLC微粒填料,如硅胶和以硅胶为基质的键合相、氧化铝、有机聚合物微球(包括离子交换树脂)等的粒度通常在3μm、5μm、7μm、以及10μm。采用的固定相粒度甚至可以达到1μm,而制备色谱所采用的固定相粒度通常大于10μm。HPLC填充柱效的理论值可以达到50000/m~160000/m理论板,一般采用100-300mm的柱长可满足大多数样品的分析
色谱法测催化剂比表面积实验报告
化工专业实验报告 实验名称:色谱法测定固体催化剂的表面积 实验人员:同组人: 实验地点:天大化工技术实验中心606室 实验时间:2015年4月17号 年级;专业;组号;学号指导教师: 实验成绩: 天津大学化工技术实验中心印制 一.实验目的 1.掌握用流动吸附色谱法测定催化剂比表面积的方法。 2.通过实验了解BET多层吸附理论在测定比表面积方面的应用。 二.实验原理 催化剂的表面积是其重要的物性之一。表面积的大小直接影响催化剂的效能。因此在催化剂研究、制造和应用的过程中,测定催化剂的表面积是十分重要的。固体催化剂表面积的测定方法较多。经典的BET法,由于设备复杂、安装麻烦,应用受到一定限制。气相色谱的发展,为催化剂表面积测定提供了一种快速方法。色谱法测定催化剂固体表面积,不需要复杂的真空系统,不接触水银,操作和数据处理较简单,因此在实验室和工厂中得到了广泛应用。色谱法测固体比表面积是以氮为吸附质、以氢气或氦气作为载气,二者按一定的比例通入样品管,当装有待测样品的样品管浸入液氮时,混合气中的氮气被样品所吸附,而载气不被吸附,He-N2混气或H2-N2混气的比例发生变化。这时在记录仪上出现吸附峰。各种气体的导热系数不尽相同,氢和氦的导热系数比氮要大得多,具体各种气体的导热系数如下表1。 表1气体导热系数表 气体组分H2He Ne O2 N2 导热系数39.7 33.6 10.87 5.7 5.66
Cal/cm·sec·c°×105 同样,在随后的每个样品解吸过程中,被吸附的N2又释放出来。氮、氦气体比例的变化导致热导池与匹配电阻所构成的惠斯登电桥中A、B二端电位失去平衡,计算机通过采样板将它记录下来得到一个近似于正态分布的电位-时间曲线,称为脱附峰。最后在混合气中注入已知体积的纯氮,得到一个校正峰。根据校正峰和脱附峰的峰面积,即可计算在该相对压力下样品的吸附量。改变氮气和载气的混合比,可以测出几个氮的相对压力下的吸附量,从而可据BET公式计算表面积。BET公式: (1) 式中:P—氮气分压,Pa; P0—吸附温度下液氮的饱和蒸气压,Pa; V m—待测样品表面形成单分子层所需要的N2体积,ml; V—待测样品所吸附气体的总体积,ml;C-与吸附有关的常数 其中 V=标定气体体积×待测样品峰面积/标定气体峰面积 标定气体体积需经过温度和压力的校正转换成标准状况下的体积。以P/[V(P0-P)]对P/P0作图,可得一条直线,其斜率为(C-1)/(V m C),截距为1/(V m C),由此可得:V m=1/(斜率+截距)(2) 若知每个被吸附分子的截面积,可求出催化剂的表面积,即 S g=(V m N A A m)/(22400W)(3) 式中S g—催化剂的比表面积,m2/g; N A—阿弗加德罗常数; A m—被吸附气体分子的横截面积,其值为16.2×10-20m2; W——待测样品重量,g; BET公式的使用范围P/P0=0.05~0.35,相对压力超过此范围可能发生毛细管凝聚现象三.实验流程 本实验采用3H-2000Ⅱ型氮吸附比表面仪用色谱法测定催化剂的比表面,见图1。
高效液相色谱法(HPLC)的概述
此帖与GC版的对应,是为了让大家更好的学习和了解LC 主要内容包括: 1.高效液相色谱法(HPLC)的概述 2. 高效液相色谱基础知识介绍(1——13楼) 3. 高压液相色谱HPLC发展概况、特点与分类 4. 液相色谱的适用性 5.应用 高效液相色谱法(HPLC)的概述 以高压液体为流动相的液相色谱分析法称高效液相色谱法(HPLC)。其基本方法是用高压泵将具有一定极性的单一溶剂或不同比例的混合溶剂泵入装有填充剂的色谱柱,经进样阀注入的样品被流动相带入色谱柱内进行分离后依次进入检测器,由记录仪、积分仪或数据处理系统记录色信号或进行数据处理而得到分析结果。 由于高效液相色谱法具有分离效能高、选择性好、灵敏度高、分析速度快、适用范围广(样品不需气化,只需制成溶液即可)、色谱柱可反复使用的特点,在《中国药典》中有5 0种中成药的定量分析采用该法,已成为中药制剂含量测定最常用的分析方法。 高效液相色谱法按固定相不同可分为液-液色谱法和液-固色谱法;按色谱原理不同可分为分配色谱法(液-液色谱)和吸附色谱法(液-固色谱)等。 目前,化学键合相色谱应用最为广泛,它是在液-液色谱法的基础上发展起来的。将固定液的官能团键合在载体上,形成的固定相称为化学键合相,不易流失是其特点,一般认为有分配与吸附两种功能,常以分配作用为主。C18(ODS)为最常使用的化学键合相。 根据固定相与流动相极性的不同,液-液色谱法又可分为正相色谱法和反相色谱法,当流动相的极性小于固定相的极性时称正相色谱法,主要用于极性物质的分离分析;当流动相
的极性大于固定相的极性时称反相色谱法,主要用于非极性物质或中等极性物质的分离分析。 在中药制剂分析中,大多采用反相键合相色谱法。 系统组成: (一)高压输液系统 由贮液罐、脱气装置、高压输液泵、过滤器、梯度洗脱装置等组成。 1.贮液罐 由玻璃、不锈钢或氟塑料等耐腐蚀材料制成。贮液罐的放置位置要高于泵体,以保持输液静压差,使用过程应密闭,以防止因蒸发引起流动相组成改变,还可防止气体进入。2.流动相 流动相常用甲醇-水或乙腈-水为底剂的溶剂系统。 流动相在使用前必须脱气,否则很易在系统的低压部分逸出气泡,气泡的出现不仅影响柱分离效率,还会影响检测器的灵敏度甚至不能正常工作。脱气的方法有加热回流法、抽真空脱气法、超声脱气法和在线真空脱气法等。 3.高压输液泵 是高效液相色谱仪的关键部件之一,用以完成流动相的输送任务。对泵的要求是:耐腐蚀、耐高压、无脉冲、输出流量范围宽、流速恒定,且泵体易于清洗和维修。高压输液泵可分为恒压泵和恒流泵两类,常使用恒流泵(其压力随系统阻力改变而流量不变)。 (二)进样系统 常用六通阀进样器进样,进样量由定量环确定。操作时先将进样器手柄置于采样位置(L OAD),此时进样口只与定量环接通,处于常压状态,用微量注射器(体积应大于定量环体积)注入样品溶液,样品停留在定量环中。然后转动手柄至进样位置(INJECT),使定量环接入输液管路,样品由高压流动相带入色谱柱中。 (三)色谱柱 由柱管和填充剂组成。柱管多用不锈钢制成。柱内填充剂有硅胶和化学键合固定相。在化学键合固定相中有十八烷基硅烷键合硅胶(又称ODS柱或C18柱)、辛烷基硅烷键合硅
仪器分析色谱实验报告
高效液相色谱法测定食醋和酱油中苯甲酸钠和山梨 酸钾含量 HPLC in soy sauce and vingar sodium benzoate potassium sorber content 指导老师:张志清教授 学生姓名:敬亚娟 摘要:[目的]用高效液相色谱法测定食醋和酱油中苯甲酸钠和山梨酸钾含量。【方法】采用RP-HPLC法以Hyperclone BDS C18 柱(150×4.60 nm,5um,phenomenex)为色谱柱;流动相:甲醇:0.02mol/l 乙醇胺(20 :80);柱温25℃,流速0.8mol/min ,检测波长230nm,进样量(标准进样量:2.5 ,5 ,7.5 ,10 ,15 ul ;样品进样量:5 ul)。【结果】:食醋中的苯甲酸钠含量为127.15899ug/mol,酱油中的苯甲酸钠含量为723.60033ug/mol,未见则出山梨酸钾。 Abstract: [purpose] with high-performance liquid chromatography (HPLC) in soy sauce and vinegar and sodium benzoate sorbic acid potassium content.【 methods 】 the RP-HPLC method with Hyperclone BDS using C18 column (150 x 4.60 nm, 5 um, phenomenex) for chromatographic column; Mobile phase: methanol: 0.02 mol/l ethanol amine (20:80); The column temperature 25 ℃, velocity 0.8 mol/min, detected wavelength 230 nm, into the sample weight (standard sample quantity: 2.5, 5, 10, 15, 7.5; the samples into the sample weight ul: 5 ul). 【 results 】 : the content of sodium benzoate feed vinegar 127.15899 ug/mol, soy sauce, sodium benzoate content of
高效液相色谱法习题答案
第二十章高效液相色谱法 思考题和习题 1.简述高效液相色谱法和气相色谱法的主要异同点。 相同点:均为高效、高速、高选择性的色谱方法,兼具分离和分析功能,均可以在线检测 不同点: 2.何谓化学键合相?常用的化学键合相有哪几种类型?分别用于哪些液相色谱法中? 采用化学反应的方法将固定液键合在载体表面上,所形成的填料称为化学键合相。优点是使用过程不流失,化学性能稳定,热稳定性好,适于作梯度淋洗。 目前常用的Si-O-Si-C型键合相,按极性分为非极性,中等极性与极性三类。①非极性键合相:常见如ODS键合相,既有分配又有吸附作用,用途非常广泛,用于分析非极性或弱极性化合物;②中等圾性键合相:常见的有醚基键合相,这种键合相可作正相或反相色谱的固定相,视流动相的极性而定:③极性键合相:常用氨基、氰基键合相,用作正相色谱的固定相,氨基键合相还是分离糖类最常用的固定相。 3.什么叫正相色谱?什么叫反相色谱?各适用于分离哪些化合物? 正相色谱法:流动相极性小于固定相极性的色谱法。用于分离溶于有机溶剂的极性及中等极性的分子型物质,用于含有不同官能团物质的分离。 反相色谱法:流动相极性大于固定相极性的色谱法。用于分离非极性至中等极性的分子型化合物。 4.简述反相键合相色谱法的分离机制。 典型的反相键合色谱法是用非极性固定相和极性流动相组成的色谱体系。固定相,常用十八烷基(ODS或C18)键合相;流动相常用甲醇-水或乙腈-水。非典型反相色谱系统,用弱极性或中等极性的键合相和极性大于固定相的流动相组成。 反相键合相表面具有非极性烷基官能团,及未被取代的硅醇基。硅醇基具有吸附性能,剩余硅醇基的多寡,视覆盖率而定。对于反相色谱的分离机制 目前,保留机制还没有一致的看法,大致有两种观点,一种认为属于分配色谱,另一种认为属于吸附色谱。分配色谱的作用机制是假设混合溶剂(水十有机溶剂)中极性弱的有机溶剂吸附于非极性烷基配合基表面,组分分子在流动相中与被非极性烷基配合基所吸附的液相中进行分配。吸附色谱的作用机制可用疏溶剂理论来解释。这种理论把非极性的烷基键合相,看作是在硅胶表面上覆盖了一层键合的十八烷基的"分子毛",这种"分子毛'有强的疏水特性。当用水与有机溶剂所组成的极性溶剂为流动相来分离有机化合物时,一方面,非极性组分分子或组分分子的非极性部分,由于疏溶剂作用,将会从水中被"挤"出来,与固定相上的疏水烷基之间产生缔合作用,其结果使组分分子在固定相得到保留。另一方面,被分离物的极性部分受到极性流动相的作用,使它离开固定相,减小保留值,此即解缔过程,显然,这两种作用力之差,决定了分子在色谱中的保留行为。一般说来,固定相上的烷基配合基或被分离分子中非极性部分的表面积越
研究生液相色谱实验报告
高效液相实验报告 高乃群S080601018 实验目的 1.了解高效液相色谱仪的类型和各自的优缺点; 2.了解液相色谱仪及各检测器的工作原理 3.掌握高效液相色谱仪的操作; 4..学会高效液相实验数据的处理; 实验原理: 1.液固色谱法使用固体吸附剂,被分离组分在色谱柱上分离原理是根据固定 相对组分吸附力大小不同而分离。分离过程是一个吸附-解吸附的平衡过程。 常用的吸附剂为硅胶或氧化铝,粒度5~10μm。适用于分离分子量200~1000的组分,大多数用于非离子型化合物,离子型化合物易产生拖尾。常用于分离同分异构体。 2.液液色谱法使用将特定的液态物质涂于担体表面,或化学键合于担体表面 而形成的固定相,分离原理是根据被分离的组分在流动正相色谱法采用极性固定相(如聚乙二醇、氨基与腈基键合相);流动相为相对非极性的疏水性溶剂(烷烃类如正已烷、环已烷),常加入乙醇、异丙醇、四氢呋喃、三氯甲烷等以调节组分的保留时间。常用于分离中等极性和极性较强的化合物(如酚类、胺类、羰基类及氨基酸类等)。 反相色谱法一般用非极性固定相(如C18、C8);流动相为水或缓冲液,常加入甲醇、乙腈、异丙醇、丙酮、四氢呋喃等与水互溶的有机溶剂以调节保留时间。适用于分离非极性和极性较弱的化合物。RPC在现代液相色谱中应用最为广泛,据统计,它占整个HPLC应用的80%左右。相和固定相中溶解度不同而分离。分离过程是一个分配平衡过程。 3.离子交换色谱法固定相是离子交换树脂,常用苯乙烯与二乙烯交联形成的 聚合物骨架,在表面未端芳环上接上羧基、磺酸基(称阳离子交换树脂)或季氨基(阴离子交换树脂)。被分离组分在色谱柱上分离原理是树脂上可电离离子与流动相中具有相同电荷的离子及被测组分的离子进行可逆交换,根据各离子与离子交换基团具有不同的电荷吸引力而分离。 4.离子对色谱法根据被测组分离子与离子对试剂离子形成中性的离子对化合 物后,在非极性固定相中溶解度增大,从而使其分离效果改善。主要用于分析离子强度大的酸碱物质。 5.排阻色谱法固定相是有一定孔径的多孔性填料,流动相是可以溶解样品的溶 剂。小分子量的化合物可以进入孔中,滞留时间长;大分子量的化合物不能进入孔中,直接随流动相流出。它利用分子筛对分子量大小不同的各组分排阻能力的差异而完成分离。常用于分离高分子化合物,如组织提取物、多肽、蛋白质、核酸等。 实验设备:高效液相色谱仪
高效液相色谱法的计算方法
高效液相色谱法的计算方法 高效液相色谱法是用高压输液泵将具有不同极性的单一溶剂或不同比例的混合溶剂、缓冲液等流动相泵入装有固定相的色谱柱,经进样阀注入供试品,由流动相带入柱内,在柱内各成分被分离后,依次进入检测器,色谱信号由记录仪或积分仪记录。 1、对仪器的一般要求 所用的仪器为高效液相色谱仪。色谱柱的填料和流动相的组分应按各品种项下的规定。常用的色谱柱填料有硅胶和化学键合硅胶。后者以十八烷基硅烷键合硅胶最为常用,辛基键合硅胶次之,氰基或氨基键合硅胶也有使用;离子交换填料,用于离子交换色谱;凝胶或玻璃微球等,用于分子排阻色谱等。注样量一般为数微升。除另有规定外,柱温为室温,检测器为紫外吸收检测器。 在用紫外吸收检测器时,所用流动相应符合紫外分光光度法(附录ⅣA)项下对溶剂的要求。 正文中各品种项下规定的条件除固定相种类、流动相组分、检测器类型不得任意改变外,其余如色谱柱内径、长度、固定相牌号、载体粒度、流动相流速、混合流动相各组分的比例、柱温、进样量、检测器的灵敏度等,均可适当改变,以适应具体品种并达到系统适用性试验的要求。一般色谱图约于20分钟内记录完毕。 2、系统适用性试验 按各品种项下要求对仪器进行适用性试验,即用规定的对照品对仪器进行试验和调整,应达到规定的要求;或规定分析状态下色谱柱的最小理论板数、分离度和拖尾因子。 (1)色谱柱的理论板数(N,用于定量表示色谱柱的分离效率,简称柱效)。 在选定的条件下,注入供试品溶液或各品种项下规定的内标物质溶液,记录色谱图,量出供试品主成分或内标物质峰的保留时间tR(以分钟或长度计,下同,但应取相同单位)和半高峰宽(W h/2),按n=5.54(t R/Wh/2)2计算色谱柱的理论板数,如果测得理论板数低于各品种项下规定的最小理论板数,应改变色谱柱的某些条件(如柱长、载体性能、色谱柱充填的优劣等),使理论板数达到要求。 (2) 分离度(R)
高效液相色谱以理论常识-测含量
被分离组分在柱中的洗脱原理 Ⅱ基本概念和理论 一、基本概念和术语 1.色谱图和峰参数 ⊕色谱图(chromatogram)--样品流经色谱柱和检测器,所得到的信号-时间曲线,又称色谱流出曲线(elution profile). ⊕基线(base line)--流动相冲洗,柱与流动相达到平衡后,检测器测出一段时间的流出曲线。一般应平行于时间轴。 ⊕噪音(noise)――基线信号的波动。通常因电源接触不良或瞬时过载、检测器不稳定、流动相含有气泡或色谱柱被污染所致。 ⊕漂移(drift)基线随时间的缓缓变化。主要由于操作条件如电压、温度、流动相及流量的不稳定所引起,柱内的污染物或固定相不断被洗脱下来也会产生漂移。 ⊕色谱峰(peak)--组分流经检测器时相应的连续信号产生的曲线。流出曲线上的突起部分。正常色谱峰近似于对称性正态分布曲线(高斯Gauss曲线)。不对称色谱峰有两种:前延峰(leading peak)和脱尾峰(tailing peak ).前者少见。 ⊕拖尾因子(tailing factor,T)--T=B/A,用以衡量色谱峰的对称性。也称为对称因子(symmetry factor)或不对称因子(asymmetry factor)《中国药典》规定T应为0.95~1.05。T<0.95为前延峰,T>1.05为拖尾峰。 ⊕峰底――基线上峰的起点至终点的距离。 ⊕峰高(Peak height,h)――峰的最高点至峰底的距离。 ⊕峰宽(peak width,W)--峰两侧拐点处所作两条切线与基线的两个交点间的距离。W=4σ。⊕半峰宽(peak width at half-height,Wh/2)--峰高一半处的峰宽。W h/2=2.355σ。 ⊕标准偏差(standard deviation, σ)--正态分布曲线x=±1时(拐点)的峰宽之半。正常峰宽的拐点在峰高的0.607倍处。标准偏差的大小说明组分在流出色谱柱过程中的分散程度。σ小,分散程度小、极点浓度高、峰形瘦、柱效高;反之,σ大,峰形胖、柱效低。 ⊕峰面积(peak area,A)――峰与峰底所包围的面积。A=×σ×h=2.507σh=1.064Wh/2h 2.定性参数(保留值) ⊕死时间(dead time,t0)--不保留组分的保留时间。即流动相(溶剂)通过色谱柱的时间。在反相HPLC中可用苯磺酸钠来测定死时间。 ⊕死体积(dead volume,V0)――由进样器进样口到检测器流动池未被固定相所占据的空间。它包括4部分:进样器至色谱柱管路体积、柱内固定相颗粒间隙(被流动相占据,Vm)、柱出口管路体积、检测器流动池体积。其中只有Vm参与色谱平衡过程,其他3部粉只起峰扩展作用。为防止峰扩展,这3部分体积应尽量减小。V0=F×t0(F为流速) ⊕保留时间(retention time,tR)--从进样开始到某个组分在柱后出现浓度极大值的时间。⊕保留体积(retention volume,VR)--从进样开始到某个组分在柱后出现浓度极大值时流出溶剂的体积。又称洗脱体积。VR=F*tR . ⊕调整保留时间(adjusted retention time,tR’)--扣除死时间后的保留时间。也称折合保留时间(reduced retention time)。在实验条件(温度、固定相等)一定时,tR’只决定于组分的性质,因此,tR’(或tR)可用于定性。TR’=tR-t0 ⊕调整保留体积(adjusted retention volume,VR’)--扣除死体积后的保留体积。VR=VR-V0 或VR=F*tR’ 3.柱效参数 ⊕理论塔板数(theoretical plate number,N)用于定量表示色谱柱的分离效率(简称柱效)。 N取决于固定相的种类、性质(粒度、粒径分布等)、填充状况、柱长、流动相的种类和流速及测定柱效所用物质的性质。如果峰形对称并符合正态分布,N可近似表示为: N=(tR/σ)2=16(tR)2/W =5.54(tR/W1/2)2 W:峰宽;σ:曲线拐点处峰宽的一半,即峰高0.607处峰宽的一半。 N为常量时,W随tR成正比例变化。在一张多组分色谱图上,如果各组份含量相当,则后洗脱的峰比前面的峰要逐渐加宽,峰高则逐渐降低。 用半峰宽计算理论塔板数比用峰宽计算更为方便和常用,因为半峰宽更容易准确测定,尤其是对稍有拖尾的峰。
- 方法验证原始记录表(色谱法)
- 高效液相色谱原始记录
- 液相色谱原始记录-磺胺 (2)
- 高效液相色谱法分析(多菌灵)原始记录
- 高效液相色谱法检验记录模板
- 高效液相色谱-质谱法分析( 内吸磷)原始记录
- 高效液相色谱原始记录
- 高效液相色谱法分析(纳他霉素)原始记录
- 液相色谱分析原始记录
- 液相色谱原始记录—苯并(α)芘
- 液相色谱仪校验记录表
- 液质原始记录表
- 高效液相色谱-质谱法分析(青霉素)原始记录
- 高效液相色谱法分析(生物胺)原始记录
- 高效液相色谱-质谱法分析(灭多威)原始记录
- 高效液相色谱-质谱法分析( 杀螟丹)原始记录
- 高效液相色谱-质谱法分析(啶虫脒 )原始记录
- 高效液相色谱法分析(安赛蜜)原始记录
- 高效液相验证原始记录
- 药材检验原始记录样本